
Research Interests
Michael F. Crowley is a principal scientist in the National Renewable Energy Laboratory's (NREL's) Biosciences Center, having joined the laboratory in 2007 to develop a simulation and theory team. He leads the theory, modeling, and simulation efforts for biofuels research. He is the principal investigator for the U.S. Department of Energy's (DOE's) Bioenergy Technologies Office (BETO) project "Biochemical Process Modeling and Simulation," in which enzyme digestion, metabolic modeling, and reactor and process design are the main thrusts. He is co-principal investigator and leads the modeling thrust of C3Bio, the Energy Frontier Research Center, in which NREL is a partner with Purdue, and multiple other collaborative projects in biofuels and computational chemistry research. He and his team maintain a level of productivity that produces on the order of 15 peer-reviewed articles a year and as many presentations, and they are regarded internationally as arguably the most productive and authoritative center for biofuel and biomass simulation and theory.
Crowley is a developer of both CHARMM and Amber molecular dynamics software for biological macromolecular simulation and has contributed significant advances to highly parallel, high-performance scientific software for use on supercomputers. Until recently his team included Antti-Pekka Hynninen, who is pushing the frontier of scientific simulation using the most up-to-date computer hardware available in preparation for future computer architectures. Crowlely's group has designed and implemented new methods and algorithms specifically for biomass and biofuel-related questions.
An author on more than 130 peer-reviewed scientific articles, Crowley has given more than 200 presentations, more than 50 of those being by invitation.
Crowley received his B.S. in chemistry from Fordham University, and his M.S. and Ph.D. in physical chemistry from the University of Montana, with a short period of study at the University of Alberta. His postdoctoral fellowship was at Brandeis University with Dr. Irving Epstein. His doctoral and post-doctoral research was studying coupled chemical oscillators and coupled nonlinear differential equations, and it was the only study to date of nonforced coupled chemical reactions showing synchronization, locking, and chaotic behaviors. In his time as assistant professor at Penn State University, Crowley moved to studies of semiconductor surfaces. At West Virginia University, he applied the nonlinear dynamical theories to controlling chaos in chemical oscillators and flame fronts. In addition, at Carnegie Mellon University and the Pittsburgh Supercomputing Center, and at The Scripps Research Institute with David Case and Charles Brooks, Crowley developed highly parallel methods for simulations of bio-macromolecules.
Affiliated Research Programs
Enzyme–Microbe Development
Biomass Deconstruction & Pretreatment
Biological & Catalytic Conversion of Sugars and Lignin
Process Design, Modeling, and Economics
BioEnergy Science Center (BESC)
Center for Direct Catalytic Conversion of Biomass to Biofuels (C3Bio)
CHARMM Modernizaton and Performance
NIH-funded collaboration with Professor Charles L. Brooks III at the University of Michigan
Areas of Expertise
Biomass Degrading Enzyme Biophysics and Design
Cellulases and other biomass-active enzymes are used in the industrial conversion of biomass to its components for further upgrading to fuels and products. Research on these enzymes is focused on determining the mechanism of action, thermodynamics, and barriers in the enzymatic digestion of plant cell walls. Using molecular mechanics and dynamics with advanced sampling methods, Crowley and his team determine free energies such as binding of substrates, conformational changes, reaction profiles, and barriers, with the aim of gaining basic understanding of how the enzymes work and how they can be engineered to work more efficiently for better fuel production. The team has had successes with many cellulases and lytic polysaccharide monooxygenases, uncovering the mechanisms of their CBMs, linkers, active sites, and binding sites, and predicting mutational changes for better performance.
Cellulose Binding Domain is thermodynamically more stable on the top, hydrophobic face of cellulose.
Product (blue) binding in a Family 48 Cellobiohydrolase is bound in the pocket by amino acids (yellow), which are present all the way until their product is expelled.
Cellulose, Hemicellulose, Other Plant Cell Wall Materials
Cellulose is the most plentiful biopolymer on Earth. The research of Crowley and his team discovers the intricacies of cell-wall polymer structure and function and their interactions to form cell wall architecture. The aim has traditionally been to understand the substrate of the enzymes studied in order to improve both the enzymes and the pretreatment methods, with the long-term goal of possibly changing the cellulose that has been produced by plants. The team is expanding research to study material properties. Strong publications in cellulose structure including force field analysis, polymorph studies, decrystallization thermodynamics, and structural understanding such as how twist occurs in cellulose are being published. The team's efforts are also directed toward understanding the production of cell wall materials through synthase, transferase, and assembly modeling and simulation.
Cellulose fibril twist studied to determine source of twisting in hydrogen bonding patterns.
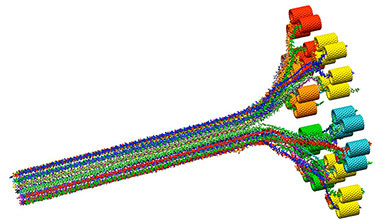
Model of cellulose fibril formation from synthase rosettes in plants.
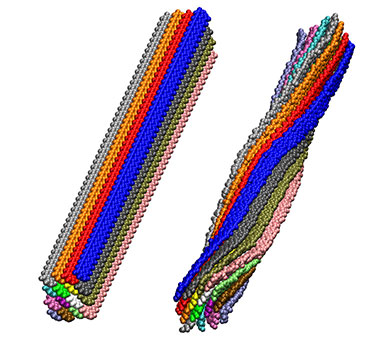
Cellulose fibril twist studied to determine source of twisting in hydrogen bonding patterns.
Cellulose fibril twist studied to determine source of twisting in hydrogen bonding patterns. This modeling will help to understand the interactions that are important in cell walls and in breaking them down to component sugars to be upgraded to fuels.
Cellulosomes
In BESC, the computational modeling has centered on the mechanisms of recalcitrance and bacterial conversion of cell wall material. One of the most fascinating and effective mechanisms found in bacteria is the cellulosome approach in which anywhere from a few to tens of enzymes and other cell-wall-active components are assembled on scaffolds, which may be tethered to the bacterium bind and act on plant cell walls to depolymerize cellulose, hemicellulose, and lignin to oligomers that can be upgraded within the bacterial cell.
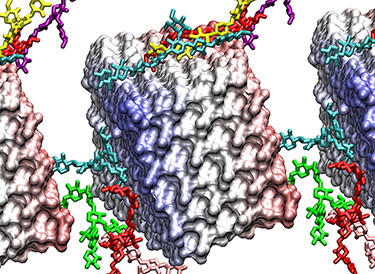
Coarse-grain model of complex cellulosome with 7 primary scaffolds on a secondary scaffold interacting with three coarse-grain cellulose fibrils. The cellulosome and its enzyme components have many cellulose-binding domains that result in the binding behavior viewed here.
Coarse-grain model of complex cellulosome with 7 primary scaffolds on a secondary scaffold interacting with three coarse-grain cellulose fibrils. The cellulosome and its enzyme components have many cellulose-binding domains that result in the binding behavior viewed here.
Sugar Upgrading Enzyme Biophysics and Design
In the Biochemical Platform of the BETO, Crowley leads the Biochemical Process Modeling and Simulation Project, which partners with experimental approaches to producing fuels and high-value products from sugars and lignin. The modeling is directed at bottlenecks in the molecular and metabolic processes involved in transport and upgrading. This includes enzyme design for selectivity, activity, pH and temperature optimization, and prediction of mutations and heterologous pathway design.
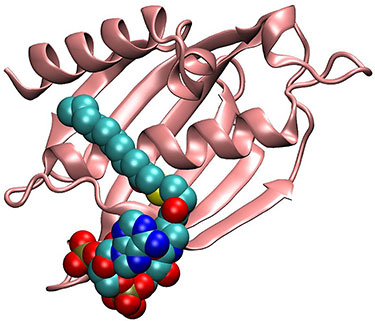
Thioesterase is a key enzyme in producing fatty acids from sugars in the pathway from sugars to diesel and aviation fuels by microbes.
Molecular Dynamics and Simulation Code Development
CHARMM and Amber code development is an integral part of Crowley's work. He has been a developer of Amber and CHARMM since 1995 and has contributed to the high-performance aspects of both and he has led efforts to modernize them. The development of the fast DOMDEC code in CHARMM was led by Crowley through the heroic efforts of Antti-Pekka Hynninen at NREL and has brought CHARMM to the level of performance of other high-performing codes carrying with it the vast and unparalleled scientific functionality and the introduction of parallel GPU implementations. This work continues with new methods of sampling that harness the immense computational power of the leadership-class machines in DOE and the XSEDE centers.
Education
Postdoctoral Fellow, Brandeis University
Ph.D., Physical Chemistry, University of Montana
M.S., Physical Chemistry, University of Montana
B.S., Chemistry, Fordham University
Featured Publications
-
"The Molecular Origins of Twist in Cellulose I-beta," Carbohydrate Polymers (2015)
-
"Predicting enzyme adsorption to lignin films by calculating enzyme surface hydrophobicity," Journal of Biological Chemistry (2014)
-
"Carbohydrate-protein interactions that drive processive polysaccharide translocation in enzymes revealed from a computational study of cellobiohydrolase processivity," Journal of the American Chemical Society (2014)
View all NREL Publications for Michael F. Crowley.
Awards and Honors
U.S. Department of Energy SciDAC Electronic Visualization Competition, Second Place Cel7A Animation (2008)
Share
